You might also like
- SpectrosDocument28 pagesSpectrosPraveen Kumar AvvaruNo ratings yet
- Analytical Methods Selection GuideDocument16 pagesAnalytical Methods Selection GuideSubhash Dhungel100% (1)
- Analytical ChemistryDocument14 pagesAnalytical ChemistryDrMd Idris100% (2)
- Intro To Electroanalytical ChemistryDocument38 pagesIntro To Electroanalytical Chemistryarun231187No ratings yet
- Applications of Uv VisibleDocument3 pagesApplications of Uv VisibleLaiba ZulfiqarNo ratings yet
- Optical SpectrosDocument7 pagesOptical SpectrosgayaxniNo ratings yet
- Analytical ChemistryDocument14 pagesAnalytical ChemistryHarrizul Rivai100% (1)
- An Introduction To Instrumental Methods PDFDocument4 pagesAn Introduction To Instrumental Methods PDFReagan Go100% (1)
- UNIT 14 Introductio To Analytical Instruments - Doc 12.5Document14 pagesUNIT 14 Introductio To Analytical Instruments - Doc 12.5NathanianNo ratings yet
- Applied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistryDocument33 pagesApplied Chemistry (UCB008) : - Instructor - Dr. Soumen Basu Associate Professor, School of Chemistry and BiochemistrySukh SindhiNo ratings yet
- Uv Visible SpectrosDocument28 pagesUv Visible Spectrosjoshishravan3003No ratings yet
- IR SpectrosDocument60 pagesIR SpectrosdeepakNo ratings yet
- Spectroscopy Analysis GuideDocument11 pagesSpectroscopy Analysis GuideAltaf Ur RehmanNo ratings yet
- Analytical Chemistry in ForensicsDocument7 pagesAnalytical Chemistry in ForensicsSergio Bugosen TannousNo ratings yet
- Voltammetry: A Look at Theory and Application: Bobby Diltz 14 March 2005Document15 pagesVoltammetry: A Look at Theory and Application: Bobby Diltz 14 March 2005tila100% (1)
- IR SpectrosDocument119 pagesIR SpectrosRojan PradhanNo ratings yet
- NMR Spectroscopy-1Document63 pagesNMR Spectroscopy-1Abhishek Kumar SinghNo ratings yet
- 1.1 Beer - Lambert's LawDocument23 pages1.1 Beer - Lambert's LawHanumant ChavanNo ratings yet
- (UV Vis) SpectrosDocument4 pages(UV Vis) SpectrosGarion Charles0% (1)
- UV-Vis Spectroscopy Group ProjectDocument50 pagesUV-Vis Spectroscopy Group ProjectVacker Guzel50% (2)
- Analytical Chemistry Lecture Pre-Limenaries MT 2ADocument10 pagesAnalytical Chemistry Lecture Pre-Limenaries MT 2AJerica Mae GabitoNo ratings yet
- Application Uv SpectrposDocument11 pagesApplication Uv SpectrposRai Shahzad NoshairNo ratings yet
- Instrumental Methods of Analysis CH6501Document13 pagesInstrumental Methods of Analysis CH6501Sûdhân HârîNo ratings yet
- ColorimetryDocument21 pagesColorimetryM.PRASAD NAIDUNo ratings yet
- Analytically ChemistryDocument24 pagesAnalytically ChemistryWaleed El-azab100% (1)
- Spectroscopy Techniques for Structure AnalysisDocument39 pagesSpectroscopy Techniques for Structure AnalysisJames Baben0% (1)
- Principles of IR SpectrosDocument6 pagesPrinciples of IR SpectrosSHEHA NUR AZZAHRA IBRAHIM -No ratings yet
- Molecular Fluorescence SpectrosDocument9 pagesMolecular Fluorescence SpectrosInga Budadoy NaudadongNo ratings yet
- 15 Spektro 01 Aas and AesDocument81 pages15 Spektro 01 Aas and AesmaudyNo ratings yet
- Mass SpectrosDocument47 pagesMass SpectrosEdward PittsNo ratings yet
- Mass SpectrosDocument19 pagesMass SpectrosRadi RamansyahNo ratings yet
- Spectrophotometry and ColorimetryDocument29 pagesSpectrophotometry and ColorimetryLea Ann Sembrano Fandida100% (2)
- Lecture 5 - Ultraviolet and Visible (UV-Vis) SpectrosDocument40 pagesLecture 5 - Ultraviolet and Visible (UV-Vis) SpectrosBelay HaileNo ratings yet
- UV SpectrosDocument25 pagesUV SpectrosokaciaNo ratings yet
- NMR Solving StrategyDocument2 pagesNMR Solving Strategysorrow Lemon100% (1)
- PharChem Lecture 3 - ACIDS and BASES (Pharmaceutical Aids and Necessities)Document65 pagesPharChem Lecture 3 - ACIDS and BASES (Pharmaceutical Aids and Necessities)Gamotkoto PharmacyNo ratings yet
- UVSpectrosDocument22 pagesUVSpectrosAbu Tareq SarkerNo ratings yet
- Introduction To Molecular Spectroscopy: By: M.Z.IqbalDocument24 pagesIntroduction To Molecular Spectroscopy: By: M.Z.IqbalMuhammad TausifNo ratings yet
- M.Sc. Chemistry Analytical Chemistry GuideDocument223 pagesM.Sc. Chemistry Analytical Chemistry GuideamitNo ratings yet
- Analytical Chemistry Basic ConceptsDocument12 pagesAnalytical Chemistry Basic ConceptsNino Jay FabrosNo ratings yet
- Role of Analytical Chemists in Industry - 2Document22 pagesRole of Analytical Chemists in Industry - 2hemakumarbNo ratings yet
- UV-VIs SpectrosDocument134 pagesUV-VIs SpectrosSaumya Prasad100% (1)
- Atomic Emission SpectrosDocument5 pagesAtomic Emission SpectrosBea Uy0% (1)
- NMR 1Document60 pagesNMR 1riyaNo ratings yet
- UV-Vis InstrumentDocument7 pagesUV-Vis InstrumentNorizzatul Akmal100% (1)
- Introduction and History of Medicinal ChemistryDocument16 pagesIntroduction and History of Medicinal Chemistryrubal guptaNo ratings yet
- Analytical ChemistryDocument26 pagesAnalytical Chemistryمحمد ناصر عليويNo ratings yet
- UV Visible SpectrosDocument8 pagesUV Visible SpectrosMahendrasinh SisodiyaNo ratings yet
- Analysis of Aspirin - Infrared (Ir) Spectroscopy and Melting Point DeterminationDocument15 pagesAnalysis of Aspirin - Infrared (Ir) Spectroscopy and Melting Point DeterminationMahmoud ElshahawyNo ratings yet
- Introduction To Infrared Spectroscopy: D. Jim Livingston Faculty of Chemistry St. John'S CollegeDocument22 pagesIntroduction To Infrared Spectroscopy: D. Jim Livingston Faculty of Chemistry St. John'S CollegeJim LivingstonNo ratings yet
- NMR SpectrosDocument32 pagesNMR SpectrosRenjitha J RNo ratings yet
- Atomic Absorption Spectroscopy: Sodium atom absorb at a wavelength of 589 nm. What is the energy difference between (in J) between the ground and excited stateDocument28 pagesAtomic Absorption Spectroscopy: Sodium atom absorb at a wavelength of 589 nm. What is the energy difference between (in J) between the ground and excited statepulkit chughNo ratings yet
- Lecture 5 Molecular SpectrosDocument97 pagesLecture 5 Molecular Spectroshoboslayer97No ratings yet
- Chromatography - The Most Versatile Method of Chemical AnalysisDocument438 pagesChromatography - The Most Versatile Method of Chemical AnalysisdnabrasilNo ratings yet
- Determining Reaction MechanismsDocument34 pagesDetermining Reaction MechanismsMahmoud AbdAllahNo ratings yet
- Introduction To UV-Vis SpectrosDocument5 pagesIntroduction To UV-Vis SpectrosShahul0% (1)
- Essays on Analytical Chemistry: In Memory of Professor Anders RingbomFrom EverandEssays on Analytical Chemistry: In Memory of Professor Anders RingbomErkki WänninenNo ratings yet
- V / F ConverterDocument8 pagesV / F ConvertercoolhemakumarNo ratings yet
- Instrument Control Uning LABVIEWDocument69 pagesInstrument Control Uning LABVIEWcoolhemakumarNo ratings yet
- Instrumental Analysis Lecture Notes IIDocument56 pagesInstrumental Analysis Lecture Notes IIcoolhemakumar100% (3)
- Mass SpectrometryDocument19 pagesMass SpectrometrycoolhemakumarNo ratings yet
- IR Spectroscopy BasicsDocument16 pagesIR Spectroscopy BasicscoolhemakumarNo ratings yet
- Interfacing To Microprocessor ControllerDocument14 pagesInterfacing To Microprocessor ControllercoolhemakumarNo ratings yet
- Interaction of EM Radiation and MatterDocument3 pagesInteraction of EM Radiation and MattercoolhemakumarNo ratings yet
- Virtual Instrumentation ArchitectureDocument25 pagesVirtual Instrumentation Architecturecoolhemakumar100% (8)
- Electromagnetic RadiationDocument2 pagesElectromagnetic RadiationcoolhemakumarNo ratings yet
- Basics of V/F and F/V ConvertersDocument11 pagesBasics of V/F and F/V ConverterscoolhemakumarNo ratings yet
- Unit 7 Flame PhotometryDocument32 pagesUnit 7 Flame PhotometryRia Agnez100% (8)
- LabVIEW IntroductionDocument89 pagesLabVIEW Introductioncoolhemakumar100% (1)
- NMR SpectrometerDocument13 pagesNMR SpectrometercoolhemakumarNo ratings yet
- Digital and Pulse-Train Conditioning: Digital I/O InterfacingDocument6 pagesDigital and Pulse-Train Conditioning: Digital I/O InterfacingcoolhemakumarNo ratings yet
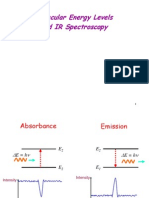
- Molecular Energy Levels and IR SpectrosDocument27 pagesMolecular Energy Levels and IR SpectroscoolhemakumarNo ratings yet
- Virtual Instrumentation SystemsDocument7 pagesVirtual Instrumentation SystemscoolhemakumarNo ratings yet
- Displacement and PositionDocument6 pagesDisplacement and PositioncoolhemakumarNo ratings yet
- Instrumental Methods of Analysis Lesson NotesDocument80 pagesInstrumental Methods of Analysis Lesson NotescoolhemakumarNo ratings yet
- Electron Spin ResonanceDocument29 pagesElectron Spin Resonancecoolhemakumar100% (2)
- Automation in DVMsDocument7 pagesAutomation in DVMscoolhemakumarNo ratings yet
- Mass SpecDocument38 pagesMass SpecRashi VermaNo ratings yet
- Energy Levels and Transitions in AtomsDocument7 pagesEnergy Levels and Transitions in AtomscoolhemakumarNo ratings yet
- NMR Spectroscopy - Short NoteDocument6 pagesNMR Spectroscopy - Short Notecoolhemakumar100% (4)
- What Is Modular InstrumentationDocument7 pagesWhat Is Modular InstrumentationcoolhemakumarNo ratings yet
- NMRDocument142 pagesNMRMaulana Senjaya SusiloNo ratings yet
- CASE ToolsDocument5 pagesCASE ToolscoolhemakumarNo ratings yet
- Mass Spectrometry - Short NoteDocument2 pagesMass Spectrometry - Short NotecoolhemakumarNo ratings yet
- Hart TutorialDocument6 pagesHart TutorialabhiklNo ratings yet
- AN713 - Controller Area Network (CAN) BasicsDocument9 pagesAN713 - Controller Area Network (CAN) BasicsRam Krishan SharmaNo ratings yet
- Tosoh CatalogDocument116 pagesTosoh Catalogsaeedazadi1352No ratings yet
- HPLCDocument8 pagesHPLCShaffan MohdNo ratings yet
- Method Development & Optimization: Selection of The HPLC Method and SystemDocument16 pagesMethod Development & Optimization: Selection of The HPLC Method and SystemBruno IndústriaNo ratings yet
- Theory of HPLC HilicDocument33 pagesTheory of HPLC HilicNguyen Duc Khanh ThoNo ratings yet
- Gas Chromatography Differences ExplainedDocument5 pagesGas Chromatography Differences ExplainedrickNo ratings yet
- Column Choices For Proteins - AgilentDocument48 pagesColumn Choices For Proteins - Agilentlejla7No ratings yet
- 15.10 Methodology LCMS IMPDocument8 pages15.10 Methodology LCMS IMPNitish TankNo ratings yet
- Metabolitos Secundarios de La Pulpa Verde de Persea AmericanaDocument6 pagesMetabolitos Secundarios de La Pulpa Verde de Persea AmericanaCarolina Salazar HurtadoNo ratings yet
- Pharmaceutical AnalysisDocument131 pagesPharmaceutical AnalysisSakhamuri Ram's100% (1)
- HPLCDocument170 pagesHPLCSai KiranNo ratings yet
- Ion Pair ChromatographyDocument9 pagesIon Pair ChromatographyLeo EspositoNo ratings yet
- HPLC - Back To BasicsDocument38 pagesHPLC - Back To Basicsmonday125No ratings yet
- Cleaning and Maintaining C-18 Columns PDFDocument5 pagesCleaning and Maintaining C-18 Columns PDFayesha2394No ratings yet
- Top 10 HPLC Column MythsDocument8 pagesTop 10 HPLC Column MythsananedallNo ratings yet
- RP HPLCDocument9 pagesRP HPLCGoutam GhoshNo ratings yet
- Basics Fundamentals of Liquid Chromatography HPLCDocument35 pagesBasics Fundamentals of Liquid Chromatography HPLCDr-Abdulsamie Hassan Alta'eeNo ratings yet
- PCI Catalog - CompressedDocument20 pagesPCI Catalog - CompressedWalid HaouasNo ratings yet
- HPLC User's Guide, 32 Karat 8.0Document134 pagesHPLC User's Guide, 32 Karat 8.0thynameisraymond100% (1)
- Difference Reverse Phase and Normal PhaseDocument8 pagesDifference Reverse Phase and Normal PhaseValeriaCusumanoNo ratings yet
- Metformin+Glibenclamide Thesis PDFDocument114 pagesMetformin+Glibenclamide Thesis PDFnari9No ratings yet
- Chromatographic Properties of Ethanol/Water Mobile Phases On Silica Based Monolithic C18Document7 pagesChromatographic Properties of Ethanol/Water Mobile Phases On Silica Based Monolithic C18Dada LimNo ratings yet
- Various Analysis Techniques For Organic Acids and Examples of Their Application. Application Note (Shimadzu)Document16 pagesVarious Analysis Techniques For Organic Acids and Examples of Their Application. Application Note (Shimadzu)Maikel Perez NavarroNo ratings yet
- Chemical constituents from Piper betle stemDocument34 pagesChemical constituents from Piper betle stemchip_daleNo ratings yet
- RP LC Column Selection-Ricardo MartinezDocument274 pagesRP LC Column Selection-Ricardo MartinezAnonymous SiO8gwwRtoNo ratings yet
- ODS Column User GuideDocument10 pagesODS Column User Guidelejosue78No ratings yet
- High Performance Liquid Chromatography: Separation and Quantification of Components in Diet Soft DrinksDocument17 pagesHigh Performance Liquid Chromatography: Separation and Quantification of Components in Diet Soft DrinksAnonymous mKkg5dNo ratings yet
- V 83 N 4 P 784Document5 pagesV 83 N 4 P 784zsoltjoooNo ratings yet
- Liquid Chromatography: Ya.I. Yashin, A.Ya. YashinDocument26 pagesLiquid Chromatography: Ya.I. Yashin, A.Ya. YashinMandu ManNo ratings yet
- ThesisDocument94 pagesThesisSarah LimNo ratings yet