You might also like
- Pathophysiology of TuberculosisDocument2 pagesPathophysiology of TuberculosisLeikkaNo ratings yet
- Obstructive Jaundice, Choledocholithiasis Calculus CholecystitisDocument36 pagesObstructive Jaundice, Choledocholithiasis Calculus CholecystitisJohn Philip M. Lacas RN100% (2)
- Case Study in KidneyDocument3 pagesCase Study in KidneyVenice VelascoNo ratings yet
- Rabies Case PresentationDocument36 pagesRabies Case PresentationViorica Gavriliță100% (1)
- BFCDocument8 pagesBFCIrene GunongNo ratings yet
- Acute Post-Streptococcal GlomerulonephritisDocument15 pagesAcute Post-Streptococcal GlomerulonephritisJeanne Marie ValesNo ratings yet
- Nursing Care Plan for Crohn's Disease ManagementDocument13 pagesNursing Care Plan for Crohn's Disease ManagementJay Jay JayyiNo ratings yet
- Patho DengueDocument3 pagesPatho DengueLindy Shane BoncalesNo ratings yet
- Course in The WardDocument4 pagesCourse in The Wardenzomarfal100% (1)
- Concept MapDocument1 pageConcept Mapadriana100% (1)
- Hypertension: Colegio de San Juan de LetranDocument13 pagesHypertension: Colegio de San Juan de LetranJenna AbuanNo ratings yet
- Drug Study: Sulodexide, Cilostazol, Metoprolol, Albumin, FurosemideDocument10 pagesDrug Study: Sulodexide, Cilostazol, Metoprolol, Albumin, FurosemideMei PayumoNo ratings yet
- Case Study ReportDocument23 pagesCase Study Reportapi-290866384No ratings yet
- Group 5 - Experiment No.10 - Culture and SensitivityDocument11 pagesGroup 5 - Experiment No.10 - Culture and SensitivityPMG BrightNo ratings yet
- Case Study 1 (Pneumonia)Document13 pagesCase Study 1 (Pneumonia)Kate EscotonNo ratings yet
- Pediatric Community Acquired Pneumonia - Moderate Risk: University of The Cordilleras College of NursingDocument22 pagesPediatric Community Acquired Pneumonia - Moderate Risk: University of The Cordilleras College of NursingJemimah AdaclogNo ratings yet
- Pathophysiology of Covid-19 Infection: What Is The Novel Coronavirus (Sars-Cov-2) Doing To Body? A Comprehensive Systematic ReviewDocument15 pagesPathophysiology of Covid-19 Infection: What Is The Novel Coronavirus (Sars-Cov-2) Doing To Body? A Comprehensive Systematic ReviewMade DeanaNo ratings yet
- Ulcerative ColitisDocument18 pagesUlcerative ColitisHoussein EL HajjNo ratings yet
- Age NCPDocument4 pagesAge NCPnj_pink081794No ratings yet
- Amoebiasis PathophysiologyDocument3 pagesAmoebiasis PathophysiologyApril CornejoNo ratings yet
- Case Report No1Document9 pagesCase Report No1Menn PetchuayNo ratings yet
- Cs AGNDocument177 pagesCs AGNMa Rafaela Rosales PalomponNo ratings yet
- Upper Gastrointestinal BleedingDocument69 pagesUpper Gastrointestinal Bleedingeliza luisNo ratings yet
- BSN4D-SG2 DM Type2Document201 pagesBSN4D-SG2 DM Type2Charisse CaydanNo ratings yet
- Nursing DiagnosisDocument5 pagesNursing DiagnosisGeovanni Rai HermanoNo ratings yet
- Part 1Document37 pagesPart 1anon_154149519No ratings yet
- Hypertension Pathophysiology and Treatment PDFDocument6 pagesHypertension Pathophysiology and Treatment PDFBella TogasNo ratings yet
- Acute Glomerulonephritis: What is it and What Causes itDocument12 pagesAcute Glomerulonephritis: What is it and What Causes itSara Sonnya Ayutthaya NapitupuluNo ratings yet
- Pathophysiology of Rheumatic Heart Disease to CardiomegalyDocument2 pagesPathophysiology of Rheumatic Heart Disease to CardiomegalyRj Avila100% (1)
- FilariasisDocument2 pagesFilariasismariaNo ratings yet
- Rabies: A Deadly Viral Disease Spread by Animal BitesDocument3 pagesRabies: A Deadly Viral Disease Spread by Animal BitesDavid CalaloNo ratings yet
- BioethicsCasesEEI 316232215 PDFDocument38 pagesBioethicsCasesEEI 316232215 PDFAman UllahNo ratings yet
- SEPTICARTHRITISDocument2 pagesSEPTICARTHRITISapi-3822433No ratings yet
- Nursing Care Plans for Pneumonia and HemorrhoidsDocument6 pagesNursing Care Plans for Pneumonia and HemorrhoidsCharlene Grace ReginoNo ratings yet
- CHF Group 3 Ncmb312 RleDocument39 pagesCHF Group 3 Ncmb312 RleMaica Lectana50% (2)
- Pneumonia Drug StudyDocument3 pagesPneumonia Drug Studyatienza02No ratings yet
- Dengue PathophysiologyDocument1 pageDengue PathophysiologyRafael Miguel Alon Protacio50% (2)
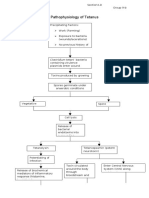
- Pathophysiology of Tetanus: Clostridium Tetani BacteriaDocument3 pagesPathophysiology of Tetanus: Clostridium Tetani BacteriaSlepy chng100% (1)
- Non-Modifiable Factor Modifiable Factor: South-East Asia, Eastern, Mediterranean, Western Pacific, and The AmericasDocument2 pagesNon-Modifiable Factor Modifiable Factor: South-East Asia, Eastern, Mediterranean, Western Pacific, and The Americaschristian quiaoitNo ratings yet
- NURSING CARE PLAN FOR RHEUMATIC HEART DISEASEDocument11 pagesNURSING CARE PLAN FOR RHEUMATIC HEART DISEASECharm TanyaNo ratings yet
- Relationship Between CKD Stage and Pulmonary Edema on Chest X-RayDocument6 pagesRelationship Between CKD Stage and Pulmonary Edema on Chest X-RayAnnisa RabbaniNo ratings yet
- Pathophysiology of HyperthyroidismDocument4 pagesPathophysiology of HyperthyroidismKitty YuffieNo ratings yet
- Facebook data privacy scandal affects 87 millionDocument1 pageFacebook data privacy scandal affects 87 millionAnne DSNo ratings yet
- Parenteral Fluid Therapy: Types of Intravenous SolutionDocument18 pagesParenteral Fluid Therapy: Types of Intravenous SolutionKathleen Joy Costales Magtanong100% (1)
- REVALIDADocument53 pagesREVALIDAMercy Anne EcatNo ratings yet
- B NCP On and Off Fever 2b ImproveddocxDocument6 pagesB NCP On and Off Fever 2b ImproveddocxKylie CatralNo ratings yet
- CholelithiasisDocument6 pagesCholelithiasismarkzamNo ratings yet
- AGE PathophysiologyDocument2 pagesAGE Pathophysiologyjosephcanlas67% (3)
- Dnrle Influenza NCPDocument3 pagesDnrle Influenza NCPEna RodasNo ratings yet
- Chronic Renal FailureDocument3 pagesChronic Renal FailureAura Salve Ildefonso AllasNo ratings yet
- College of Nursing: Cebu Normal UniversityDocument4 pagesCollege of Nursing: Cebu Normal UniversityFaye Andrea FranciscoNo ratings yet
- ParacetamolDocument2 pagesParacetamolsleep whatNo ratings yet
- The Pathophysiology of LabyrinthitisDocument2 pagesThe Pathophysiology of LabyrinthitisSurya Michael ChanceNo ratings yet
- Pathophysiology of Urinary Tract ObstructionDocument50 pagesPathophysiology of Urinary Tract ObstructionPryo UtamaNo ratings yet
- Tatz Pa ToolDocument23 pagesTatz Pa Toolian_mendoza_3No ratings yet
- PUCAN, Julienne BSN III-D - SGD - HYPO&HYPERCHLOREMIADocument10 pagesPUCAN, Julienne BSN III-D - SGD - HYPO&HYPERCHLOREMIAJulienne PucanNo ratings yet
- Nursing Care PlanDocument8 pagesNursing Care PlanVincent QuitorianoNo ratings yet
- Nephrithic Sydrome PDFDocument20 pagesNephrithic Sydrome PDFLeónGarcíaNo ratings yet
- Acute Glomerulonephritis: Causes, Symptoms and TreatmentDocument16 pagesAcute Glomerulonephritis: Causes, Symptoms and TreatmentPrabha Amandari SutyandiNo ratings yet
- Acute GlomerulonephritisDocument12 pagesAcute Glomerulonephritiskuchaibaru90No ratings yet
- Antihypertensive Drugs - Classification and SynthesisDocument14 pagesAntihypertensive Drugs - Classification and SynthesisCường NguyễnNo ratings yet
- High Risk New BornDocument36 pagesHigh Risk New BornIze C VijiNo ratings yet
- COPD A Multifactorial Systemic DiseaseDocument9 pagesCOPD A Multifactorial Systemic DiseaseNjala SankhulaniNo ratings yet
- CV Disease Drug StudyDocument11 pagesCV Disease Drug StudyMaria Francheska OsiNo ratings yet
- Angiotensin II Receptor AntagonistDocument22 pagesAngiotensin II Receptor AntagonistaakshitNo ratings yet
- Pelvic Floor Distress Inventory Questionnaire - Short Form 20Document2 pagesPelvic Floor Distress Inventory Questionnaire - Short Form 20Dana Ysabelle Ibarra100% (1)
- English Role Play Script for Nursing StudentsDocument4 pagesEnglish Role Play Script for Nursing StudentsIefana Fihtreey TaezZkeyaNo ratings yet
- Stillbirth FinalDocument21 pagesStillbirth Finalzahirah nurNo ratings yet
- Guide to Calcium, Phosphate, and Parathyroid Hormone DisordersDocument7 pagesGuide to Calcium, Phosphate, and Parathyroid Hormone DisordersGregNo ratings yet
- Nursing Care Plan Guide for Holistic Patient CareDocument23 pagesNursing Care Plan Guide for Holistic Patient CareRej PanganibanNo ratings yet
- Efficacy and Safety of Rabeprazole in Children.20Document10 pagesEfficacy and Safety of Rabeprazole in Children.20Ismy HoiriyahNo ratings yet
- Lecture - 3 Properties of Cardiac MuscleDocument35 pagesLecture - 3 Properties of Cardiac MuscleMRM7MDNo ratings yet
- PD2Document15 pagesPD2DrShobhit RajNo ratings yet
- Tugas Remedial PaperDocument7 pagesTugas Remedial PaperFujiNo ratings yet
- Tips For The NDECCDocument2 pagesTips For The NDECCMagda Jakubowska-EwiczNo ratings yet
- Makalah Basic Life SupportDocument22 pagesMakalah Basic Life SupportKustian PramuditaNo ratings yet
- Novus Knowledge Webinar - Feed Milling For Sustainable Food Production - Feed Mill Biosecurity - Dr. Charles Stark PDFDocument43 pagesNovus Knowledge Webinar - Feed Milling For Sustainable Food Production - Feed Mill Biosecurity - Dr. Charles Stark PDFaradhya bhatiya0% (1)
- Literature ReviewDocument3 pagesLiterature Reviewapi-534368223No ratings yet
- Hyponatremic Dehydration PathoDocument4 pagesHyponatremic Dehydration PathoENo ratings yet
- Discharge PlanningDocument1 pageDischarge PlanningChyNo ratings yet
- Table 5. Mrs. L's Braden ScoreDocument1 pageTable 5. Mrs. L's Braden ScoreEfryl MaeNo ratings yet
- DYSTOCIA TITLEDocument14 pagesDYSTOCIA TITLEsailor MoonNo ratings yet
- Nursing Responsibilities Adverse Effect Indication / Contraindication Mechanism of Action Drug Name IndicationDocument1 pageNursing Responsibilities Adverse Effect Indication / Contraindication Mechanism of Action Drug Name IndicationOmar IzzoNo ratings yet
- Promoting Hygiene Through Complete BathingDocument12 pagesPromoting Hygiene Through Complete BathingErlangga PratamaNo ratings yet
- Drug Name Mecahnism of Action Indication Side Effects Nursing Responsibilities Generic Name: Brand Name: Classification: - Body As A Whole: - DuringDocument2 pagesDrug Name Mecahnism of Action Indication Side Effects Nursing Responsibilities Generic Name: Brand Name: Classification: - Body As A Whole: - DuringhahahaNo ratings yet
- Cytomegalovirus in Primary ImmunodeficiencyDocument9 pagesCytomegalovirus in Primary ImmunodeficiencyAle Pushoa UlloaNo ratings yet
- Implant Surgery Complications - Etiology and TreatmentDocument10 pagesImplant Surgery Complications - Etiology and TreatmentStephanie JaramilloNo ratings yet
- 50 Prometric MCQ For GPDocument10 pages50 Prometric MCQ For GPDrkhslid8971% (7)
- 1 Quarter Examination in Tle 7Document3 pages1 Quarter Examination in Tle 7MARK FERNANDEZNo ratings yet
- E Diabetes CareDocument48 pagesE Diabetes CareRuwan ManjulaNo ratings yet